Articles
HPO: the importance of clinical coding in variant interpretation

One of the most prominent issues in bioinformatics is coping with an enormous amount of biological data in order to provide good quality information. It is commonly known that a considerable effort comes from genomic variants interpretation, however several other datasets have proved to be challenging, so far.
Clinical data regarding human phenotypic abnormalities, for instance, represent a powerful resource for diagnostic purposes and research applications. In fact, thousands of phenotype descriptions have been collected by geneticists and other specialists over the years.
The initial problem with computational analysis of such data was the lack of standardized vocabulary of phenotypic abnormalities characterizing human diseases. Another issue was the use of free texts in phenotype descriptions, which made data mining quite complex.
The Human Phenotype Ontology (HPO) was first introduced in 2008 with the aim of facilitating this process.
Generally speaking, ontologies are a powerful tool in computer science: they can be defined as collections of concepts and their synonyms, as well as of logical connections (e.g. “is-a”) between such concepts. The application of ontologies to human phenotypes significantly reduced concepts ambiguities and allowed scientists to easily gather information from clinical data.
The Human Phenotype Ontology is structured in subontology (Phenotypic abnormality, Clinical modifier, Mode of inheritance, Past medical history, Blood group and Frequency) and each phenotypic trait is described with standard terms associated with an identifier code.
Moreover, a list of synonyms is linked to the same code in order to facilitate the convergence of different searches. Every term has a logical connection, it is arranged in a directed acyclic graph (Figure 1) and it is connected by is-a (subclass-of) edges, such that a term represents a more specific or limited instance of its parent term(s).
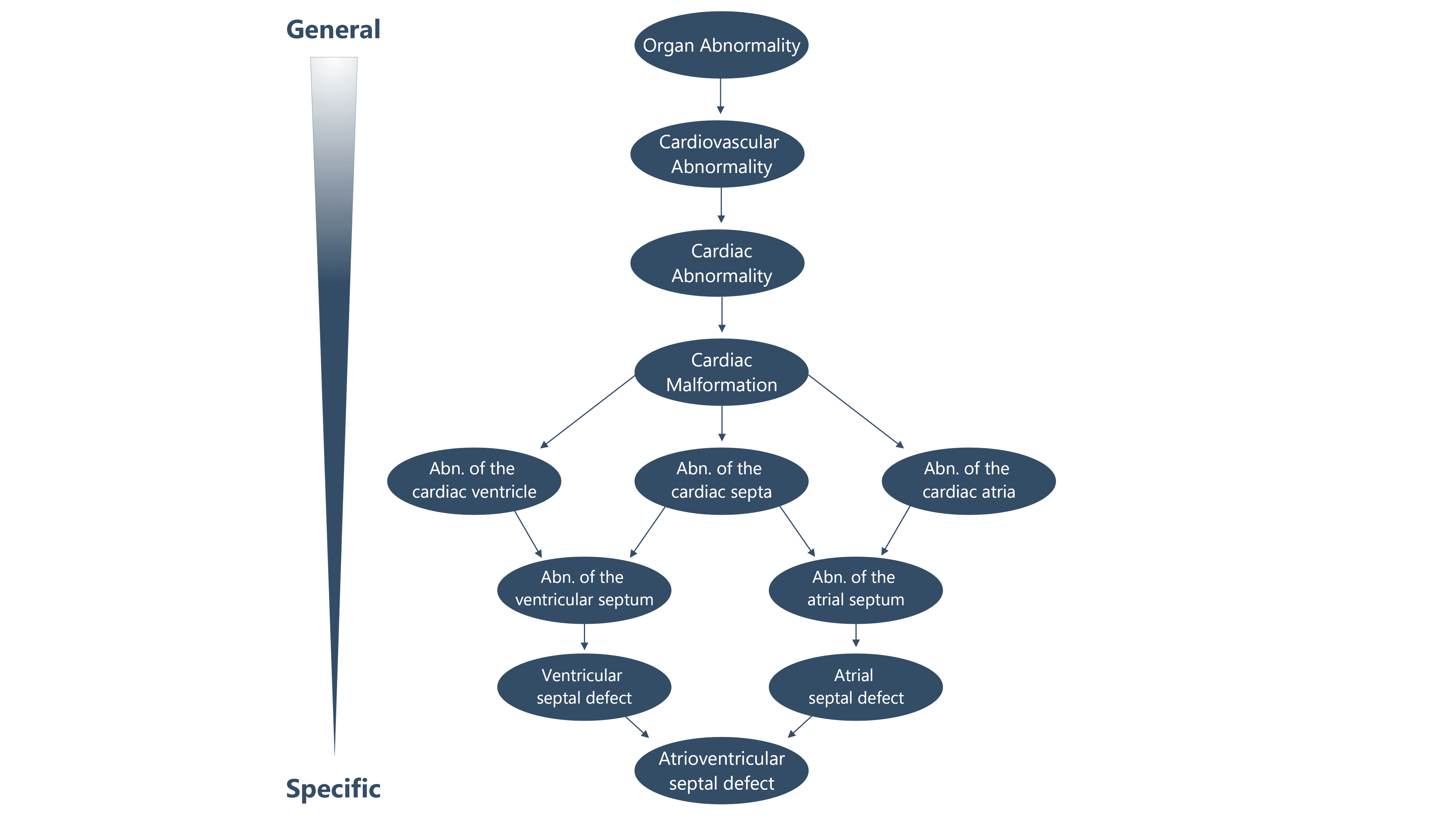
Figure 1. An example of the directed acyclic graph structure of the HPO database.
There is a clearly established pecking order among the phenotypic traits, which can always be traced back to a specific organ or a body part. Moreover, such standard terms have been used to describe a large number of genetic diseases defined by established resources such as the Online Mendelian Inheritance in Man (OMIM), Decipher and Orphanet: these descriptions have been called HPO annotations (HPOAs).
Modifiers can further expand the HPOAs, providing additional information on the age of onset, the affected sex, the related genes or the frequencies of the traits. It is also possible to distinguish whether the annotated HPO term is a major diagnostic criterion for a certain disease or a poorly characteristic symptom.
According to the database (February 2021), the HPO contains more than 16000 terms and over 200000 annotations [1, 2, 3].
The richness and the unambiguity of the HPO database allow broad research and clinical diagnostics applications. For example, researchers are able to visualize the human phenome and to analyze randomized phenotypic networks, which may lead to the discovery of interesting patterns. On the other hand, clinical experts could easily type the phenotypes of interest into the database and find the annotated diseases [4].
The most promising use for HPO, though, regards genome/exome analysis for diagnostics purposes. After sequencing, HPO terms related to the patient’s phenotype can be used to pinpoint potential pathogenic variants. This step, which relies on the prior association of the HPO terms to the pathological variants, simplifies the interpretation of genomic data to a large extent.
However, it has to be taken into account that an a priori phenotype-based analysis on a genome or exome data, might undermine the capacity to identify the disease-causing variants. This might happen when the relationships between a certain variant (or gene) and the phenotypic abnormalities are still not clear (hence not well codified), or when patient’s phenotypes are not well defined, resulting in adding noise (distant terms) and imprecision (not specific terms) to the process. Moreover, certain phenotypes are difficult to detect or are likely to appear later in time. Prenatal screening tests, for instance, are conducted when most of the phenotypic abnormalities are not present yet.
Despite these drawbacks, the use of the HPO database is a must-have in genomic interpretation softwares.
EnGenome’s eVai platform, for instance, allows for retrospectively use HPO terms by navigating the ontology tree and choosing the optimal terms to finally pinpoint pathogenic variants of interest in a phenotype-driven fashion.
Written by Vanessa Foti
[1] Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, Danis D, Balagura G, Baynam G, Brower AM, Callahan TJ, Chute CG, Est JL, Galer PD, Ganesan S, Griese M, Haimel M, Pazmandi J, Hanauer M, Harris NL, Hartnett MJ, Hastreiter M, Hauck F, He Y, Jeske T, Kearney H, Kindle G, Klein C, Knoflach K, Krause R, Lagorce D, McMurry JA, Miller JA, Munoz-Torres MC, Peters RL, Rapp CK, Rath AM, Rind SA, Rosenberg AZ, Segal MM, Seidel MG, Smedley D, Talmy T, Thomas Y, Wiafe SA, Xian J, Yüksel Z, Helbig I, Mungall CJ, Haendel MA, Robinson PN, 2021. The Human Phenotype Ontology in 2021. Nucleic Acids Research, 49 (Issue D1), pp. D1207–D1217.
[2] Hpo.jax.org. 2021. Human Phenotype Ontology. [online] Available at: https://hpo.jax.org/app/help/introduction
[3] Robinson P. and Mundlos S., 2010. The Human Phenotype Ontology. Clinical Genetics, 77(6), pp.525-534.
[4] Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S., 2008. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. American Journal of Human Genetics, 83 (5), pp. 610-5.